An Alternative Traceback Method for Nussinov's RNA Folding Algorithm
Nussinov's algorithm takes a given sequence of RNA and determines the
the most stable secondary structure for that sequence based on the
assumption that the more base pairs a structure has, the more stable
the structure will be[1]. The algorithm can be divided into two steps:
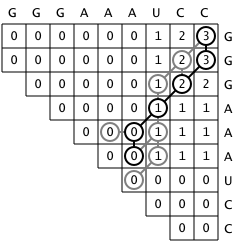
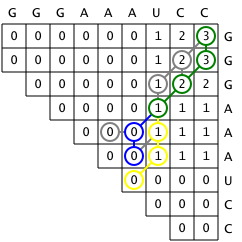
First, a matrix M is created such that any cell Mij
represents the maximum number of base pairs that a structure
consisting of bases i through j could have. The matrix is filled
incrementally by sweeping diagonally and filling each upper diagonal
using values from the previous diagonal. That is, each substructure
is scored and extended from a single base to the entire sequence so that
the score of the optimal structure for the entire sequence is in the final
cell in the upper right hand corner of the matrix.
Second, it is necessary to determine which bases are paired in this structure,
which requires going backwards from the upper right corner through the matrix
and recording which bases are paired. An important detail is that
for any given sequence, there are often multiple structures that
are considered to be optimal by the algorithm[2].
Though there are various enhancements that can be made to bring the algorithm
more in line with actual structures by modifying how the substructures are scored,
the focus of this report is on how an alternate traceback algorithm can be used
to obtain better results from Nussinov's algorithm that are normally discarded.
Specifically, the new algorithm incorporates the following assumptions:
Longer stems (consecutive base pairs) are more stable than shorter stems
A single loop or bulge is more stable than one split in two by a base pair in the middle
Generally speaking, the algorithm attempts to choose a traceback path that will
extend opened loops and extend stems when possible while still acting within the
constraints of the original algorithm. In pseudocode, the new traceback algorithm
can be described as:
last_direction = NONE
while tb_stack is not empty:
pop(tb_stack, i,j)
if i >= j:
continue
if γ(i+1,j-1) + δi,j == γ(i,j):
push(candidate_stack, DIAGONAL, i+1,j-1)
if γ(i,j-1) == γ(i,j):
push(candidate_stack, LEFT, i,j-1)
if γ(i+1,j) == γ(i,j):
push(candidate_stack, DOWN, i+1,j)
if candidate_stack is empty:
last_direction = NONE
for k from i < k < j:
if γ(i,k) + γ(k+1,j) == γ(i,j):
push(tb_stack, k+1,j)
push(tb_stack, i,k)
break
else:
while candidate_stack is not empty:
pop(candidate_stack, d, m,n)
if d == last_direction:
push(tb_stack, m,n)
if d == DIAGONAL:
record(i,j)
break
else if nothing pushed onto tb_stack:
push(tb_stack, m,n)
In the canonical implementation of the traceback step, whenever there are
multiple structures that are equivalent in terms of number of base-pairs
the first structure that works is chosen because the algorithm does not care
about anything besides the number of base-pairs, so any structure with the same
number of base pairs as the optimal one will do. However, this ignores important
information that can lead it to choose an unstable structure over a more stable one.
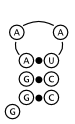
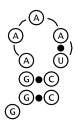
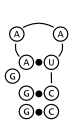
Below in figure 2 are three possible structures as predicted by Nussinov's algorithm.
All three meet the constraints of the algorithm and have the maximal three base pairs.
However, these structures are not equally stable. Figure 2a is decidedly the most stable
of the three. It forms a three base pair long stem and a three base hairpin loop (which
is more stable than a shorter hairpin loop because the angle is less sharp). Figure 2b
is rather unstable and improbable because it requires two adjacent bases to be paired,
which would require bending the bases in a nonsensical way[1].
Figure 2c is not particularly stable, but is reasonable; it is nearly identical to the
structure in figure 2a, except that it introduces a small bulge loop into the stem, which
would make it less stable.
As noted on this web page,
the structure and traceback in figure 10.9 of Biological Sequence Analysis (reproduced here as figure 1 and figure 2a respectively) are
inconsistent with the pseudocode description of the standard traceback algorithm given.
The structure is arguably the most stable of the possible foldings that Nussinov's algorithm
can predict, but is different from the one that the traceback algorithm described in the book
would choose, which is shown above in figure 2b.[3].
This alternate version of the traceback algorithm yields the correct traceback corresponding to
the optimal structure in figure 2a.
References
Durbin R, Eddy S, Krogh A, Mitchinson G. Biological Sequence Analysis. New York: Cambridge University Press. 1998.